















 {{item.name}}会员
{{item.name}}会员

Circulation | 第四军医大学西京医院重磅:METTL4介导的线粒体DNA甲基化在心脏衰竭中的调控作用
Highlights
1. HF时心肌细胞内的METTL4和mtDNA 6mA水平上升,METTL4导致线粒体功能障碍
2. 敲除或沉默METTL4可以改善心肌细胞的线粒体功能
3. 激活的p53能够增加METTL4表达,进而影响mtDNA 6mA水平
近日,“Circulation”(IF=35.5)上发表了一篇题为“Rectifying METTL4-Mediated N6-Methyladenine Excess in Mitochondrial DNA Alleviates Heart Failure”的文章。这篇文章探讨了心脏衰竭(HF)的分子机制,特别是线粒体DNA(mtDNA)上N6-甲基腺苷的异常甲基化如何导致心脏功能障碍。

研究背景介绍
N6-甲基腺苷(6mA)是一种表观遗传修饰,指在DNA分子的腺嘌呤碱基上发生的甲基化。这种修饰在原核生物中普遍存在,而在哺乳动物中,6mA被认为是一种新型的线粒体DNA修饰。
心脏衰竭(HF)是一种临床综合征,其特征是心脏泵血功能不足,无法满足身体的需求。它是多种心脏疾病的严重和终末阶段,常见原因包括冠状动脉疾病、高血压、心肌病和心脏瓣膜病。
METTL4是一种甲基转移酶,它在线粒体中催化6mA的生成。METTL4的功能异常与多种疾病的发生发展有关,包括心脏疾病。
研究思路分析

研究技术路线图
01心脏衰竭与METTL4表达的相关性
①检测HF成鼠心肌细胞线粒体DNA(mtDNA)中的5-甲基胞嘧啶(5mC)和6mA的整体水平。发现与正常心肌细胞相比,非缺血性(AngII/PE诱导)和缺血性(心肌梗死后再灌注)小鼠模型心肌细胞的mtDNA 6mA水平显著上升,而5mC水平无显著差异。
②此外,成熟心肌细胞的mtDNA 6mA水平随生理成熟而下降,但在HF时又显著增加,呈现类似新生儿的表观遗传特征。METTL4作为6mA的甲基转移酶,主要在线粒体中定位,而非核中,其表达模式与心脏衰竭发展过程中的胎儿标志基因同步上升。
③构建携带小鼠Mettl4基因的AAV9载体,并在心肌细胞中特异性过表达METTL4。发现METTL4的过表达导致心肌细胞mtDNA的6mA水平异常升高。而在没有经历任何病理压力的情况下,这些小鼠小鼠已经显示出左室功能的显著损害。
④此外,METTL4过表达还导致了心肌细胞线粒体结构和功能的异常,包括线粒体形态的异常、呼吸链复合体活性的降低和mtDNA编码的呼吸链基因mRNA水平的整体下调。
02METTL4对线粒体功能的影响及其机制
①通过在成年小鼠心肌细胞中过表达METTL4,发现这特异性地增加了心肌细胞线粒体DNA中的6mA水平,而非核DNA。这种过表达导致了线粒体电子传递链(ETC)复合物的蛋白水平降低和ETC基因mRNA表达的减少,进而影响了线粒体的功能。
②METTL4通过催化mtDNA启动子区域的6mA形成,抑制了转录起始复合物的组装,从而阻碍了mtDNA的转录和复制。此外,一个METTL4甲基转移酶活性缺失的突变体未能影响mtDNA的6mA水平和ETC基因的表达,证实了METTL4的6mA甲基化活性在其抑制作用中的必要性。
③研究显示,METTL4的mRNA和蛋白水平在心肌细胞成熟后降低,但在HF发展过程中重新表达。研究发现,METTL4基因启动子中存在一个高度保守的p53转录因子结合序列,并证实了p53与Mettl4启动子的相互作用,p53可以直接促进Mettl4基因的转录。
④在成年小鼠心肌细胞中,p53的激活通过AngII/PE刺激或Nutlin-3a药物激活,导致METTL4表达和mtDNA 6mA水平的增加,在心力衰竭的小鼠心脏中,也观察到了p53的激活。
03METTL4作为治疗靶点的潜力
①利用CRISPR/Cas9系统,创建了Mettl4fl/fl小鼠品系,并与表达心肌特异性的MerCreMer小鼠杂交,成功诱导了成年心肌细胞中Mettl4基因的条件性敲除(cKO)。经他莫昔芬诱导后,Mettl4 cKO小鼠的心肌细胞中METTL4的mRNA和蛋白表达显著降低,证实了敲除的有效性和特异性。
②在AngII/PE的长期灌注下,Mettl4 cKO小鼠显示出更好的心脏功能保护。此外,Mettl4 cKO小鼠还显示出更少的病理心脏肥大,心肌间质纤维化程度降低,以及肺水肿减轻。而AngII/PE刺激导致的mtDNA 6mA水平上升被特异性敲除Mettl4基因所阻断,且Mettl4 cKO小鼠在AngII/PE刺激下保持了线粒体功能的完整性。
③开发了两种含有靶向METTL4的shRNA的AAV9-cTNT载体。通过植入渗透泵连续灌注AngII/PE诱导心力衰竭的小鼠,随机接受AAV9-cTNT-shRNA或对照载体的静脉注射。结果表明,METTL4的沉默显著降低了心脏组织中METTL4的蛋白和mRNA水平,改善了心脏功能和结构,减少了心脏肥大和心肌间质纤维化,并降低了心力衰竭标志基因的mRNA表达水平。
④此外,METTL4的沉默还保持了线粒体呼吸功能和ETC基因的mRNA及蛋白表达。表明抑制心肌细胞中的METTL4表达可以恢复功能失调的心肌线粒体并改善不同原因引起的心力衰竭小鼠的心脏性能。
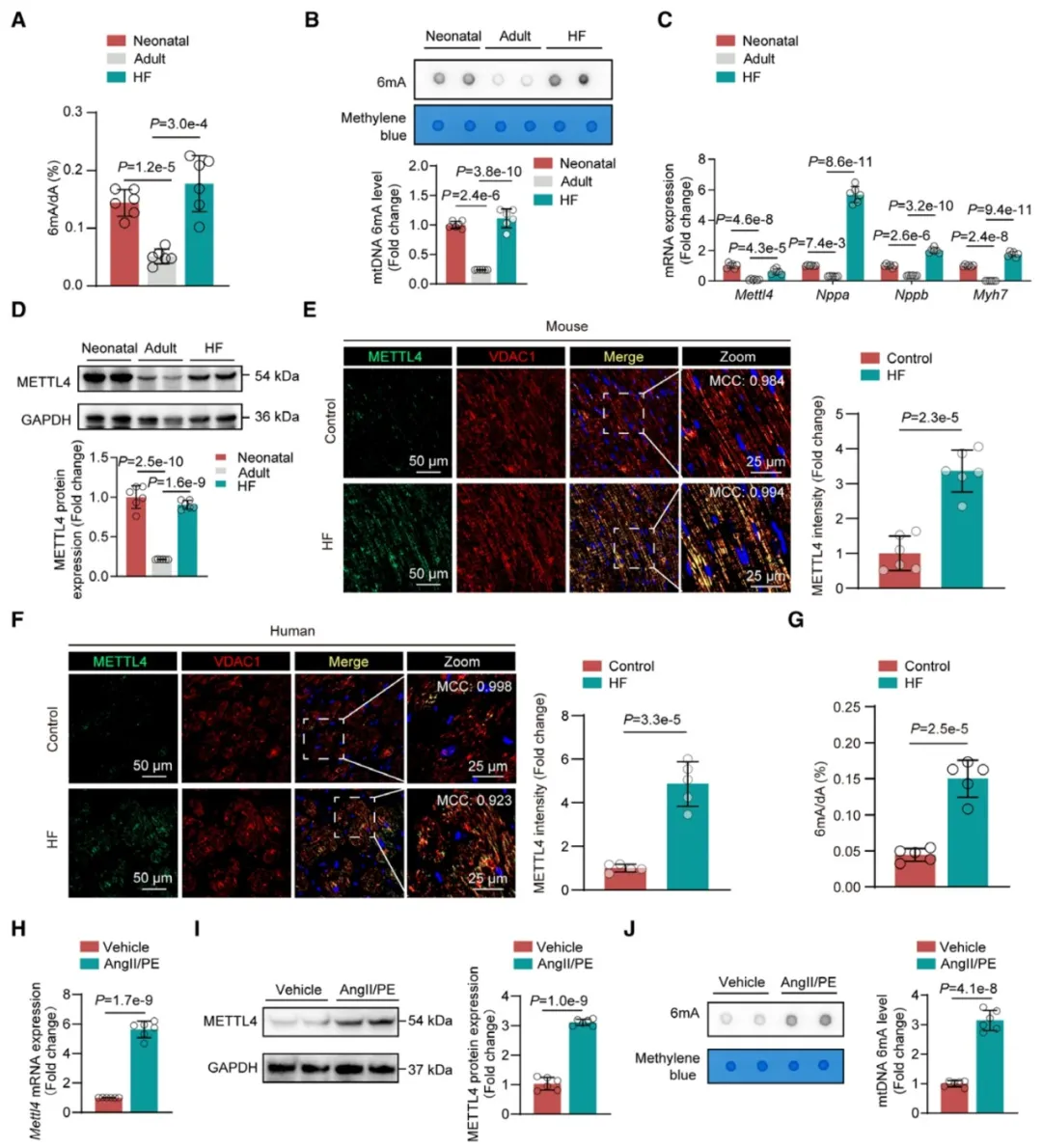
图1. 在衰竭的成人心肌细胞中mtDNA的6mA含量增加,代表了向类似新生儿状态的表观遗传学转变。
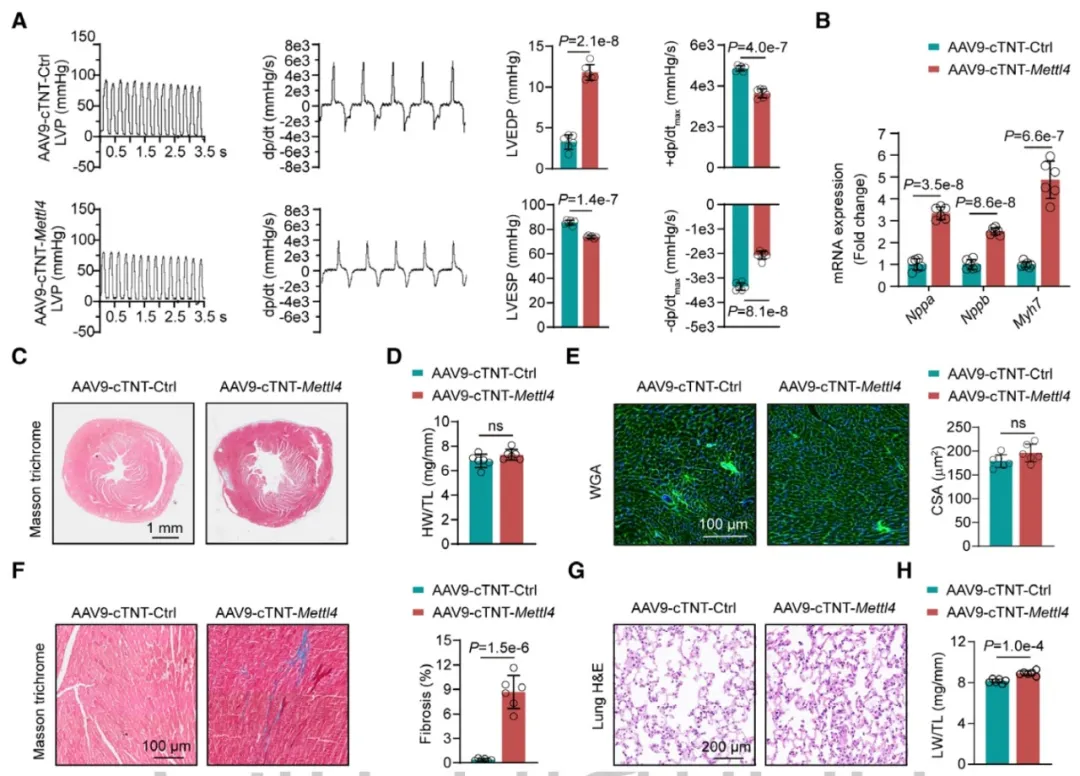
图2. 心肌细胞特异性METTL4过表达诱导自发性心力衰竭表型
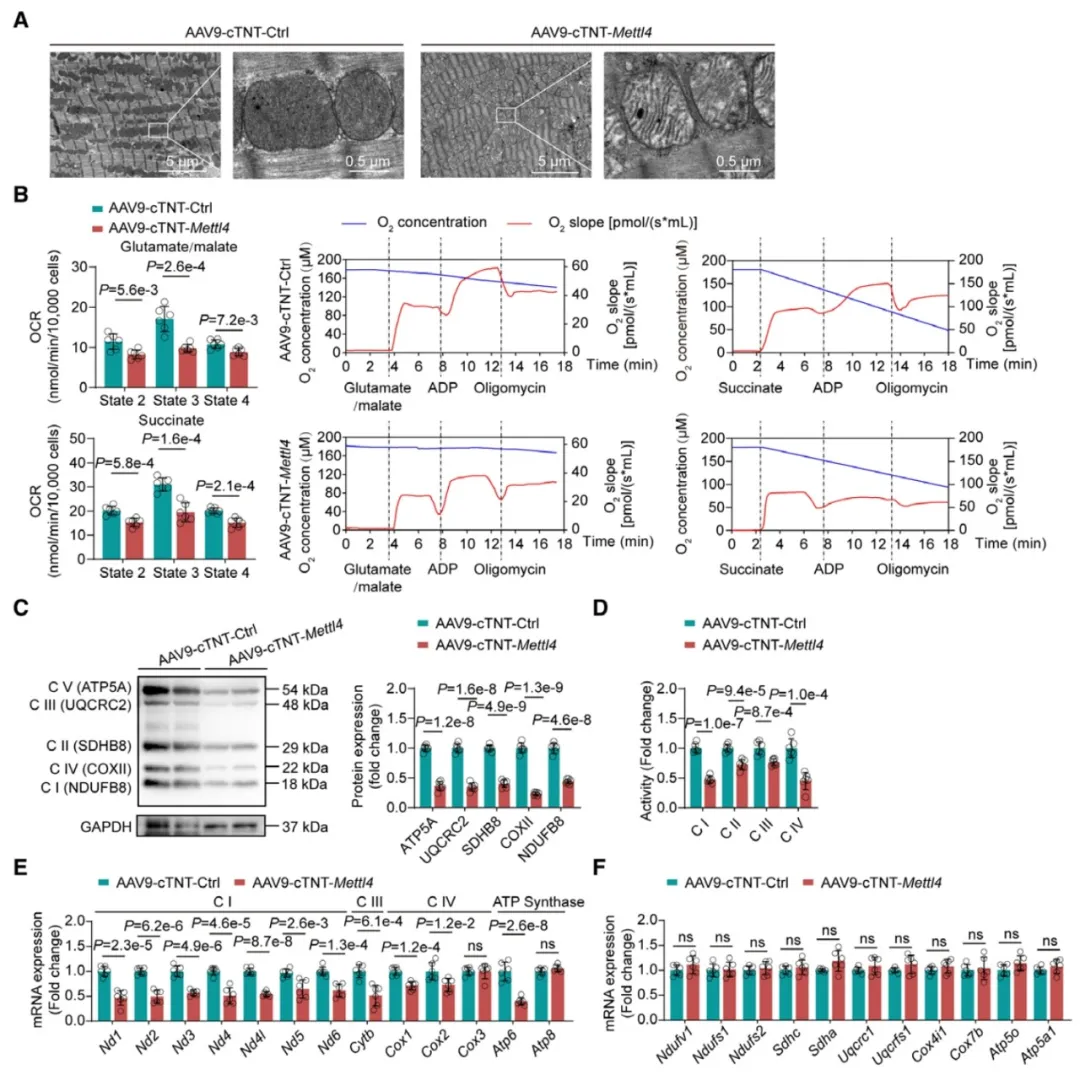
图3. 心肌细胞特异性METTL4过表达导致线粒体结构和功能障碍
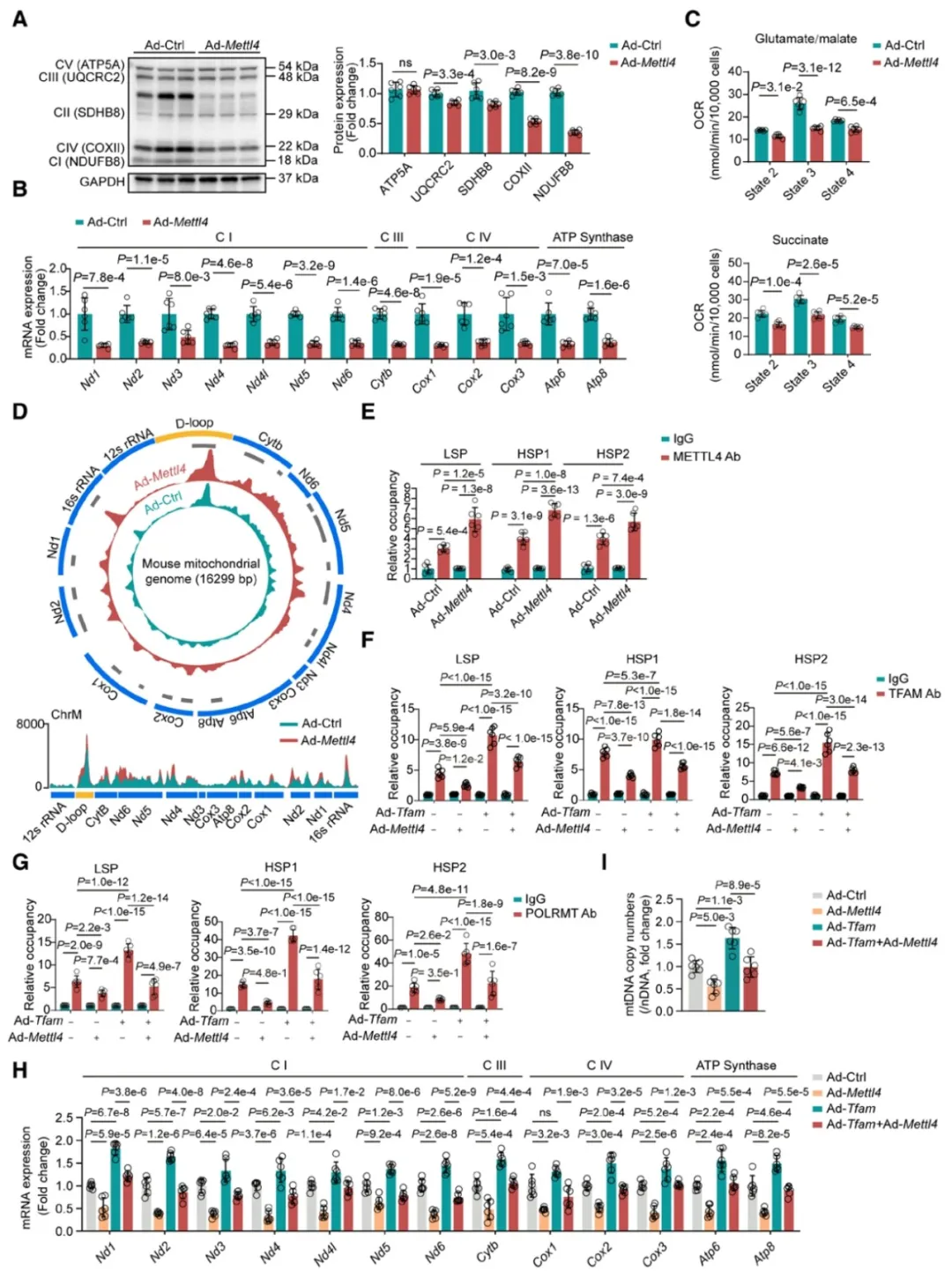
图4. METTL4通过催化mtDNA启动子区域的6mA抑制mtDNA转录
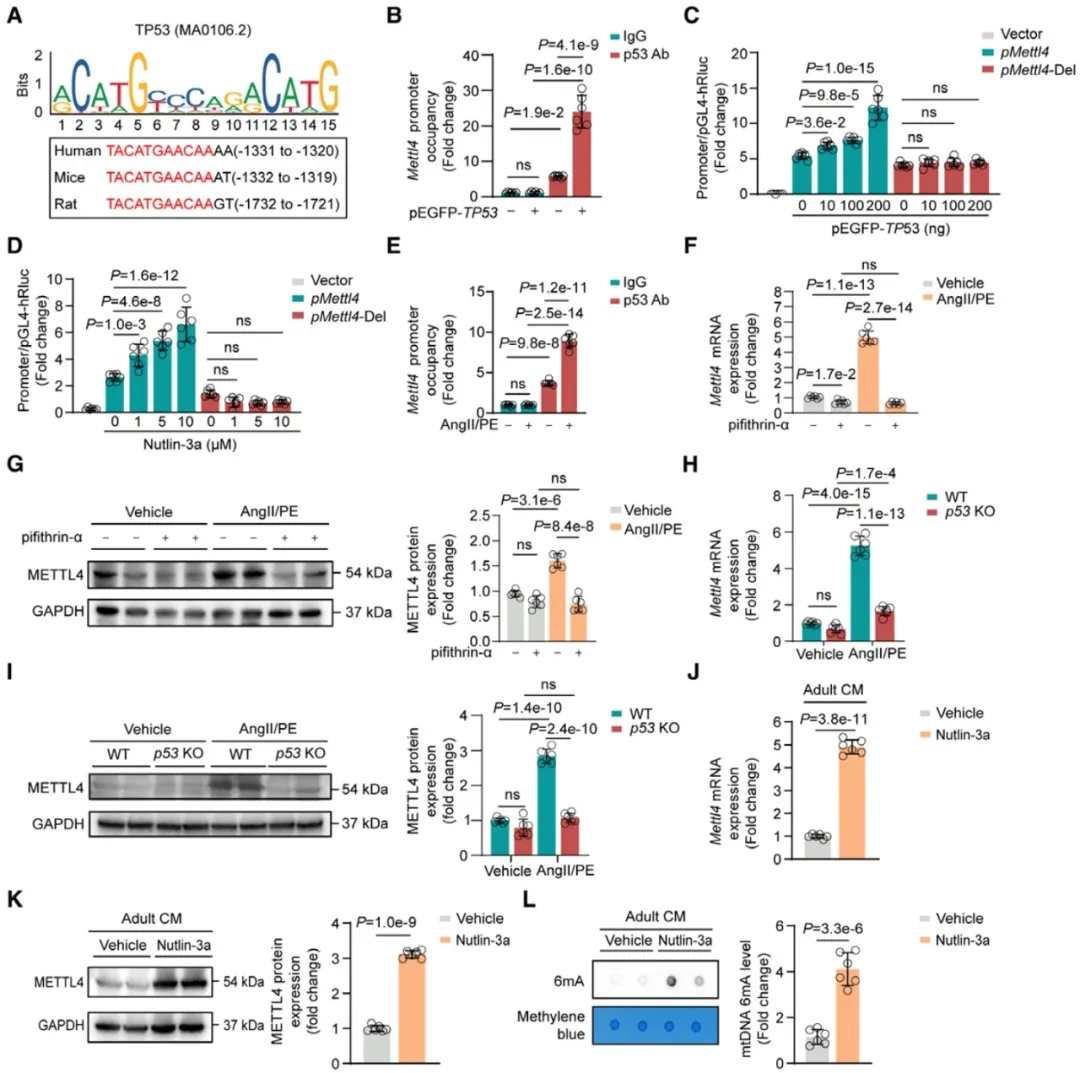
图5. 激活的p53在成人心肌细胞中直接启动Mettl4基因的转录,实现对mtDNA 6mA修饰的核调控。
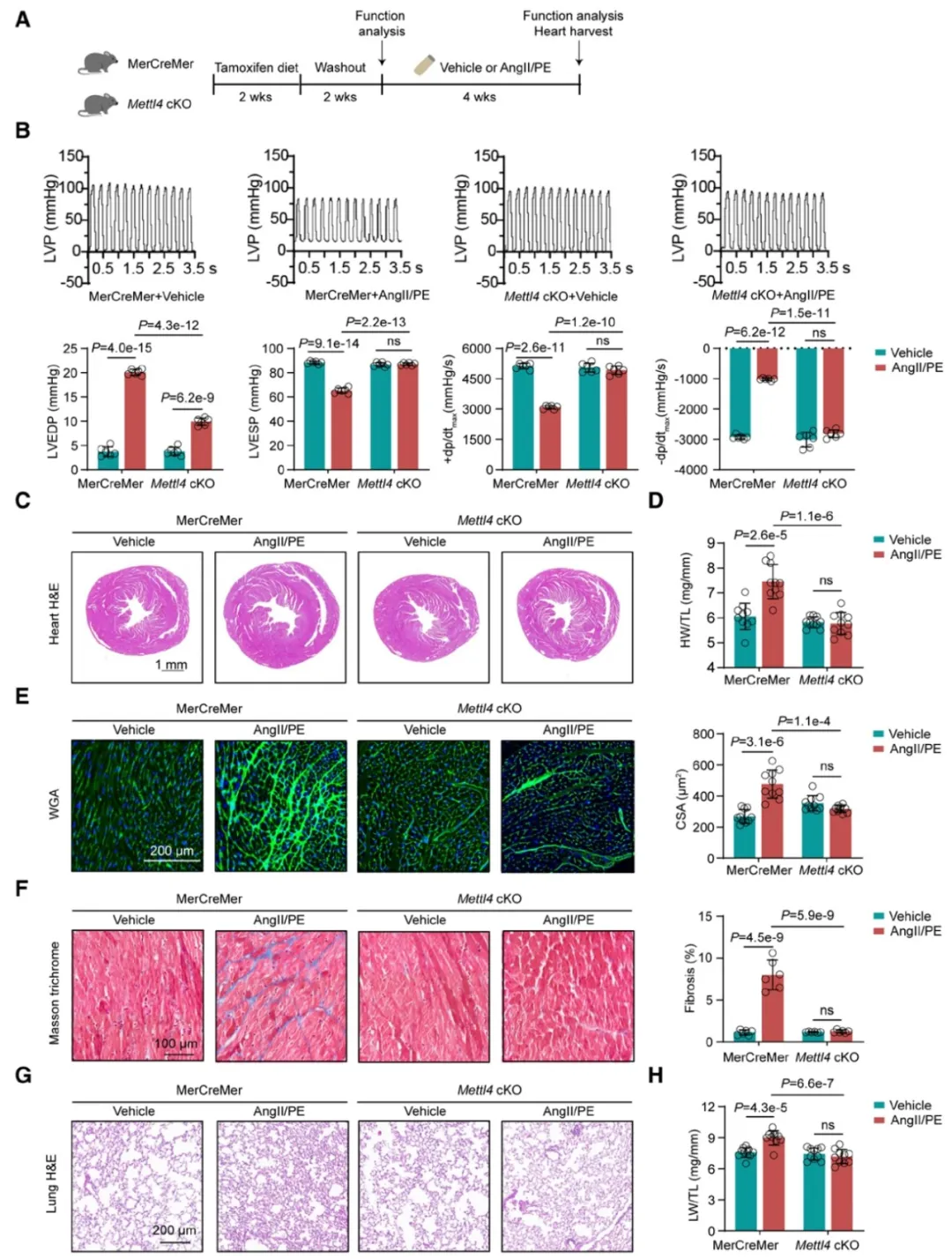
图6. 心肌细胞特异性敲除Mettl4基因保护心脏功能,防止心力衰竭的发展
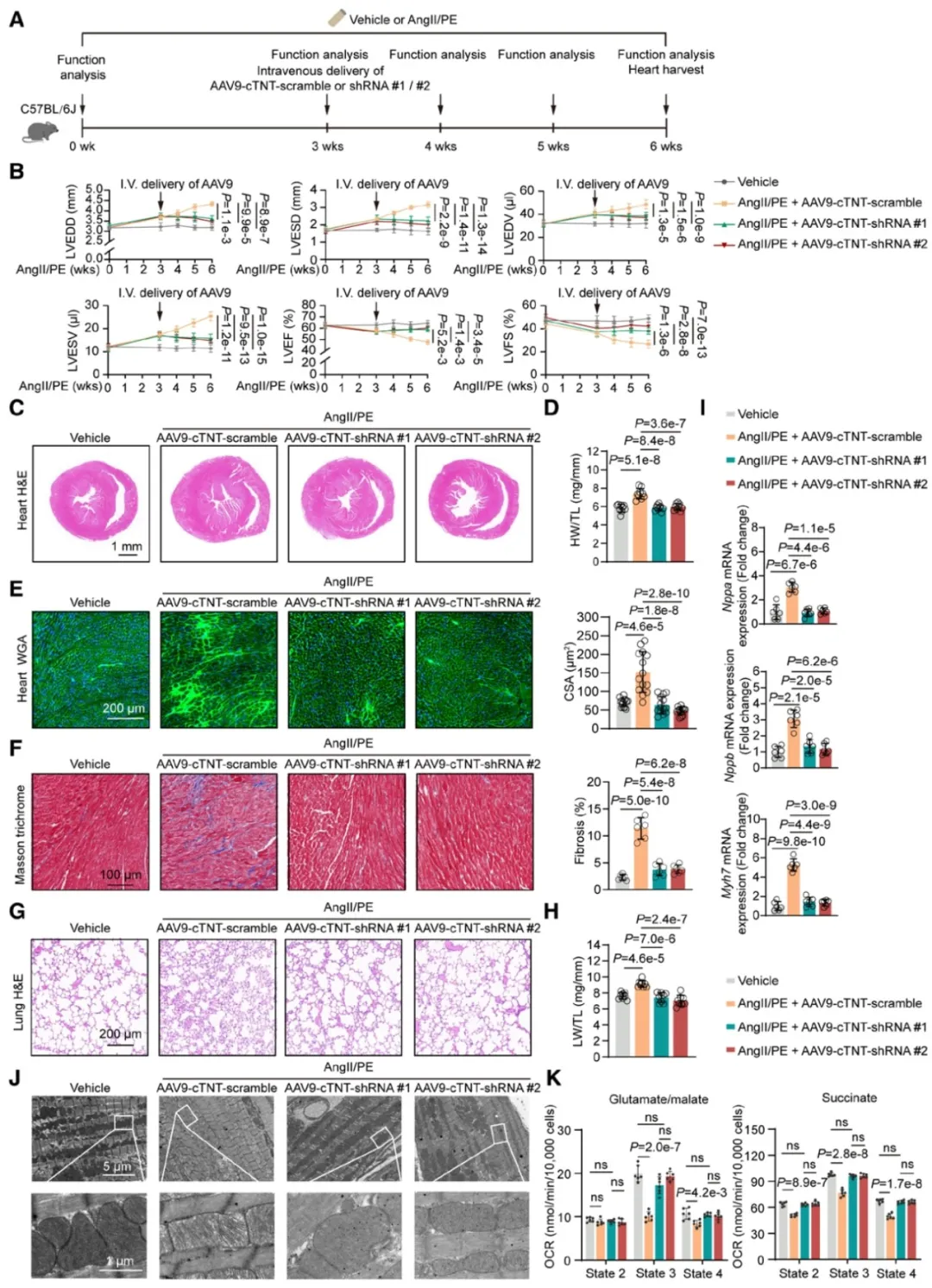
图7. 在已有心力衰竭中,心肌细胞特异性沉默METTL4恢复功能失调的心肌线粒体,改善心脏性能
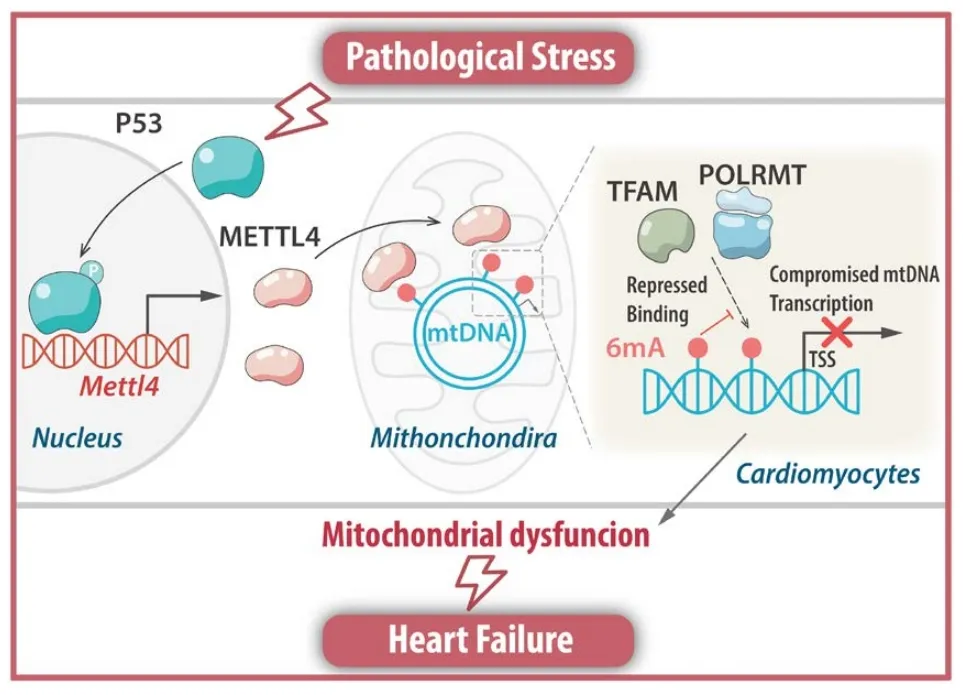
图8. Abstract
结论与讨论
本文揭示了METTL4介导的线粒体DNA 6mA修饰在心脏衰竭中的关键作用。在心脏衰竭的心肌细胞中,METTL4表达上升,导致mtDNA 6mA水平增加,进而干扰线粒体功能,诱发心脏功能障碍。通过敲除METTL4或降低其表达,能够恢复线粒体功能,减轻心脏衰竭症状,为心脏衰竭治疗提供了新的策略。
在未来,研究需进一步探索METTL4在心脏发展和疾病中的详细作用机制,以及mtDNA 6mA修饰对心脏功能影响的长期效应。此外,研究METTL4抑制剂的开发和应用,以及在不同心脏病患者中的治疗效果和安全性,将是实现临床转化的关键步骤。