















 {{item.name}}会员
{{item.name}}会员

Circulation | 北京大学研究揭示RIPK3/RAGE/CaMKII信号通路在心脏缺血再灌注损伤中的作用机制
Highlights
1. AMI接受PCI患者血浆RIPK3水平与主要不良心血管事件密切相关
2. 细胞外RIPK3是I/R的关键危险相关分子模式,与RAGE结合激活CaMKII,诱导细胞死亡和炎症
3. 细胞外RIPK3及其相关信号通路可能是治疗I/R及相关并发症的潜在靶点
近日,“Circulation”(IF=35.5)上发表了一篇题为“Extracellular RIPK3 Acts as a Danger-Associated Molecular Pattern to Exaggerate Cardiac Ischemia/Reperfusion Injury”的文章。这篇文章探讨了细胞外RIPK3在心脏缺血再灌注损伤中的作用。

研究背景介绍
损伤相关分子模式(DAMPs)是细胞受损或应激时释放的内源性分子,能够激活免疫系统并引发炎症反应。
受体相互作用蛋白激酶3(RIPK3)是调节细胞坏死和炎症的关键分子。
心脏缺血/再灌注(I/R)损伤是指在心脏缺血后恢复血流供应时,反而引发的一系列病理生理过程,这些过程可导致心肌细胞的进一步损伤。
钙/钙调蛋白依赖性蛋白激酶II(CaMKII)是一种在心脏中发挥关键作用的激酶。它在心肌细胞中的活性增加与心脏缺血/再灌注损伤有关,可能通过影响心肌细胞的电生理特性和收缩功能而参与损伤过程。
研究思路分析

研究技术路线图
01RIPK3与心脏I/R损伤的关联
①在急性心肌梗死(AMI)患者接受经皮冠状动脉介入(PCI)治疗的研究中,发现PCI后血浆中RIPK3水平的升高与患者的主要不良心血管事件(MACEs)风险增加有关,包括心血管死亡、心力衰竭再入院和血管事件。通过Kaplan-Meier分析,血浆RIPK3水平较高的患者显示出随时间增加的MACEs风险。此外,动物实验也支持了这些临床观察结果。
②通过模拟I/R损伤的实验,发现心肌细胞是细胞外RIPK3的主要来源。此外,RIPK3的释放主要是通过一个依赖于细胞内Ca2+浓度升高和SNARE蛋白Syt7的主动分泌过程,而非细胞损伤导致的被动泄漏。
02RIPK3的生物学效应
①在小鼠心脏I/R模型手术前10分钟以及再灌注后每天给予重组人RIPK3蛋白(rh-RIPK3)或RIPK3抗体处理。结果显示,rh-RIPK3的处理显著增加了心肌梗死的大小,恶化了心脏功能,并增加了心肌细胞死亡。相反,使用RIPK3抗体阻断细胞外RIPK3则显著减少了心肌梗死的大小,改善了心脏功能,并减少了心肌细胞死亡。表明细胞外RIPK3在I/R诱导的心肌梗死、心脏功能障碍和心肌细胞死亡的发展中起因果作用。
②通过使用缺氧/复氧(H/R)的体外心肌细胞模型模拟体内I/R,研究发现rh-RIPK3显著加剧了心肌细胞的死亡和炎症反应,而RIPK3抗体治疗则减轻了这些效应。此外,rh-RIPK3也增加了RAW264.7细胞中LPS诱导的炎症反应,这种效应同样可以通过RIPK3抗体阻断。在体内心脏I/R损伤模型中,rh-RIPK3处理加重了炎症因子表达和巨噬细胞浸润,而RIPK3抗体处理则减轻了这些炎症反应。表明,循环中的RIPK3在心脏I/R损伤引起的全身性炎症中起着关键作用。
③在I/R损伤中,研究表明,RIPK3加剧了内皮细胞的炎症反应和细胞凋亡,导致内皮功能障碍,这可能对微血管功能产生负面影响。通过使用rh-RIPK3和RIPK3抗体处理,发现rh-RIPK3能够加剧内皮细胞的炎症和功能障碍,而RIPK3抗体则能减轻这些效应。此外,rh-RIPK3还损害了人动脉的内皮依赖性血管舒张功能,这一影响通过RIPK3抗体得到了逆转。
03RIPK3的作用机制和临床相关性
①假设RIPK3作为损伤相关分子模式(DAMPs)通过与模式识别受体(PRRs)相互作用诱导炎症和细胞死亡。对多种PRRs进行筛选,发现RIPK3与RAGE结合。进一步研究发现,RAGE敲减显著减轻了rh-RIPK3诱导的心肌细胞炎症和死亡,并抑制了RAW264.7细胞中rh-RIPK3培养后的IL-1β和IL-6表达,以及人类主动脉内皮细胞在rh-RIPK3刺激下的内皮损伤和细胞死亡。在野生型(WT)和Ager基因敲除(KO)小鼠的I/R损伤模型中,用rh-RIPK3处理后,发现rh-RIPK3引起的心脏损伤和炎症在Ager KO小鼠中大大减轻。表明,RAGE是细胞外RIPK3的受体,并在RIPK3介导的细胞病理中发挥重要作用。
②研究发现,CaMKII在RIPK3诱导的心肌损伤中起关键作用。在小鼠I/R模型中,rh-RIPK3注射显著增强了CaMKII的自磷酸化,这一效应可以通过抗RIPK3治疗减轻。使用CaMKII抑制剂ruxolitinib可以减轻由rh-RIPK3引起的过度炎症和细胞死亡。在CaMKII-δ敲除小鼠中,rh-RIPK3处理未能加剧心肌细胞死亡或炎症。此外,RAGE敲减可以减轻rh-RIPK3引起的CaMKII过度激活。
③研究还发现,rh-RIPK3孵育增加了心肌细胞中β1肾上腺素能受体(β1AR)与RAGE的相互作用,并提高了细胞内钙离子浓度,这一过程可以通过β1AR抑制剂来阻断。表明,细胞外RIPK3与RAGE结合,促进其与β1AR的异二聚体化,从而增加细胞内Ca2+,进而引发CaMKII激活和下游病理反应。
④从PCI后6小时的患者血液中分离出人类外周血单个核细胞(PBMCs),并与健康对照组一起检测CaMKII的磷酸化水平。发现,PCI后患者血浆中的RIPK3浓度显著更高,且PBMCs中CaMKII的磷酸化水平也增加。最重要的是,CaMKII磷酸化水平与血浆RIPK3浓度呈正相关,这表明人类细胞外RIPK3浓度与细胞内CaMKII磷酸化之间存在关联。
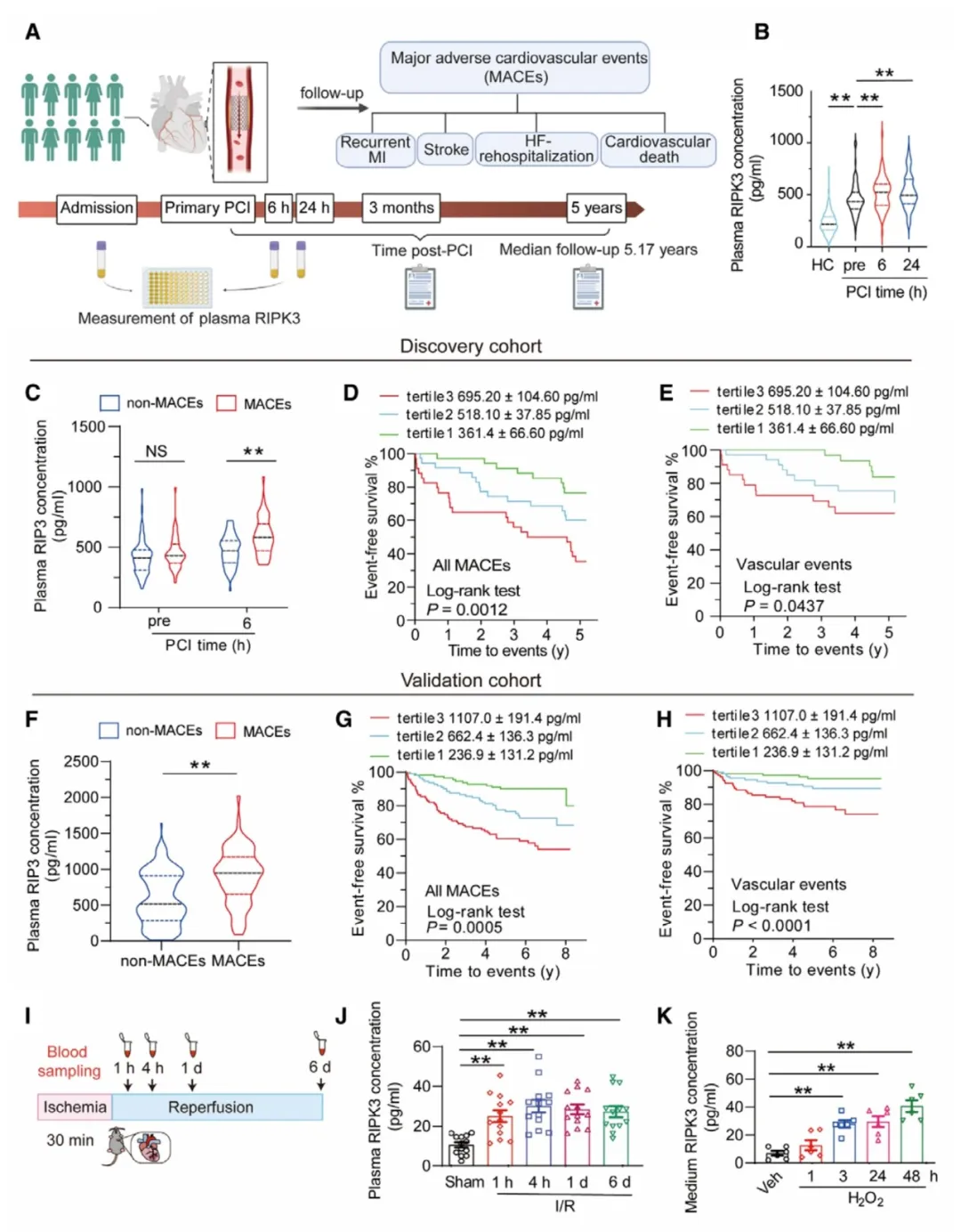
图1. 细胞外RIPK3与接受PCI治疗的AMI患者的主要不良心血管事件正相关。
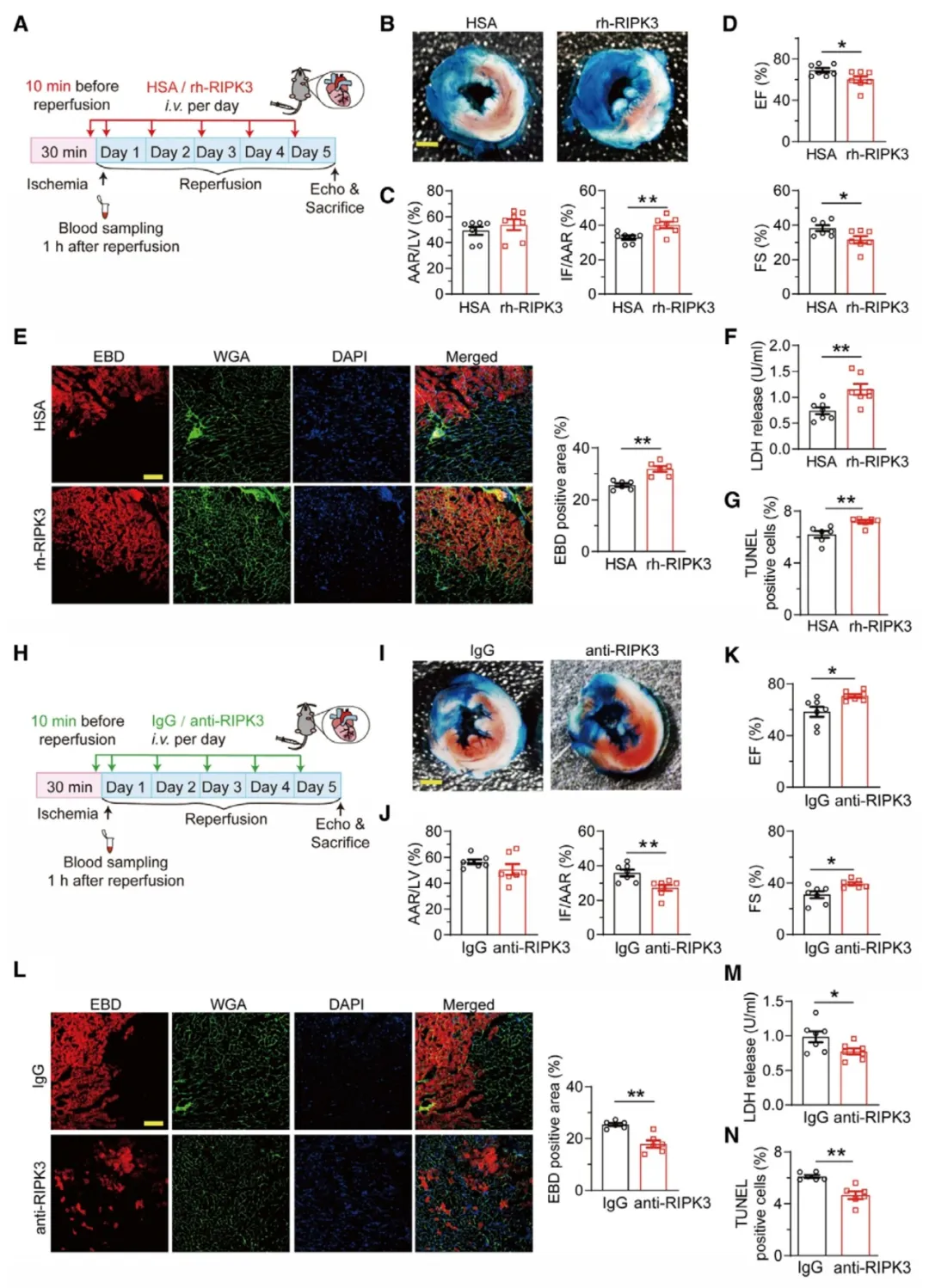
图2.细胞外RIPK3在心肌I/R损伤中发挥因果作用。
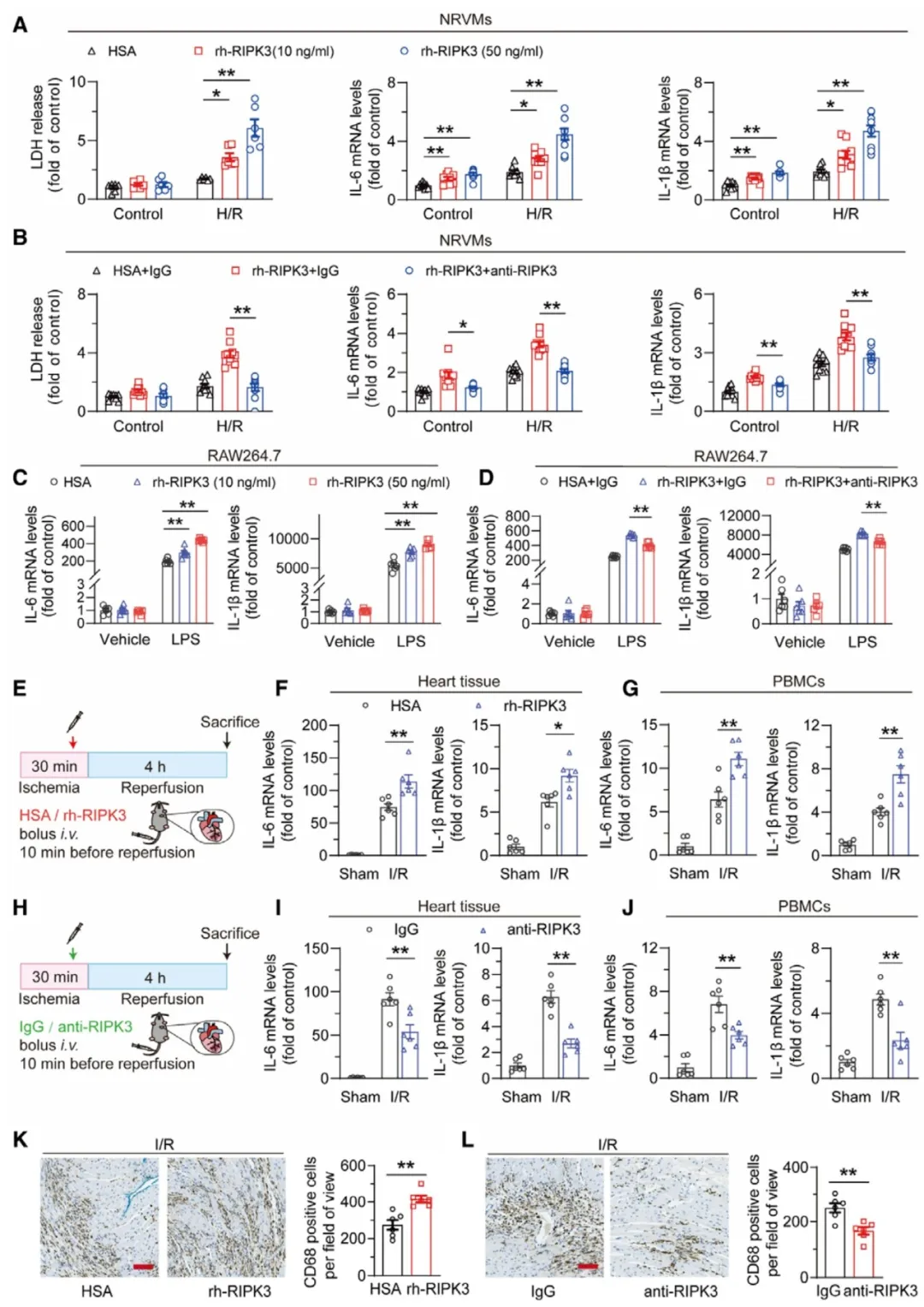
图3. 细胞外RIPK3夸大了I/R诱导的细胞死亡和炎症反应。
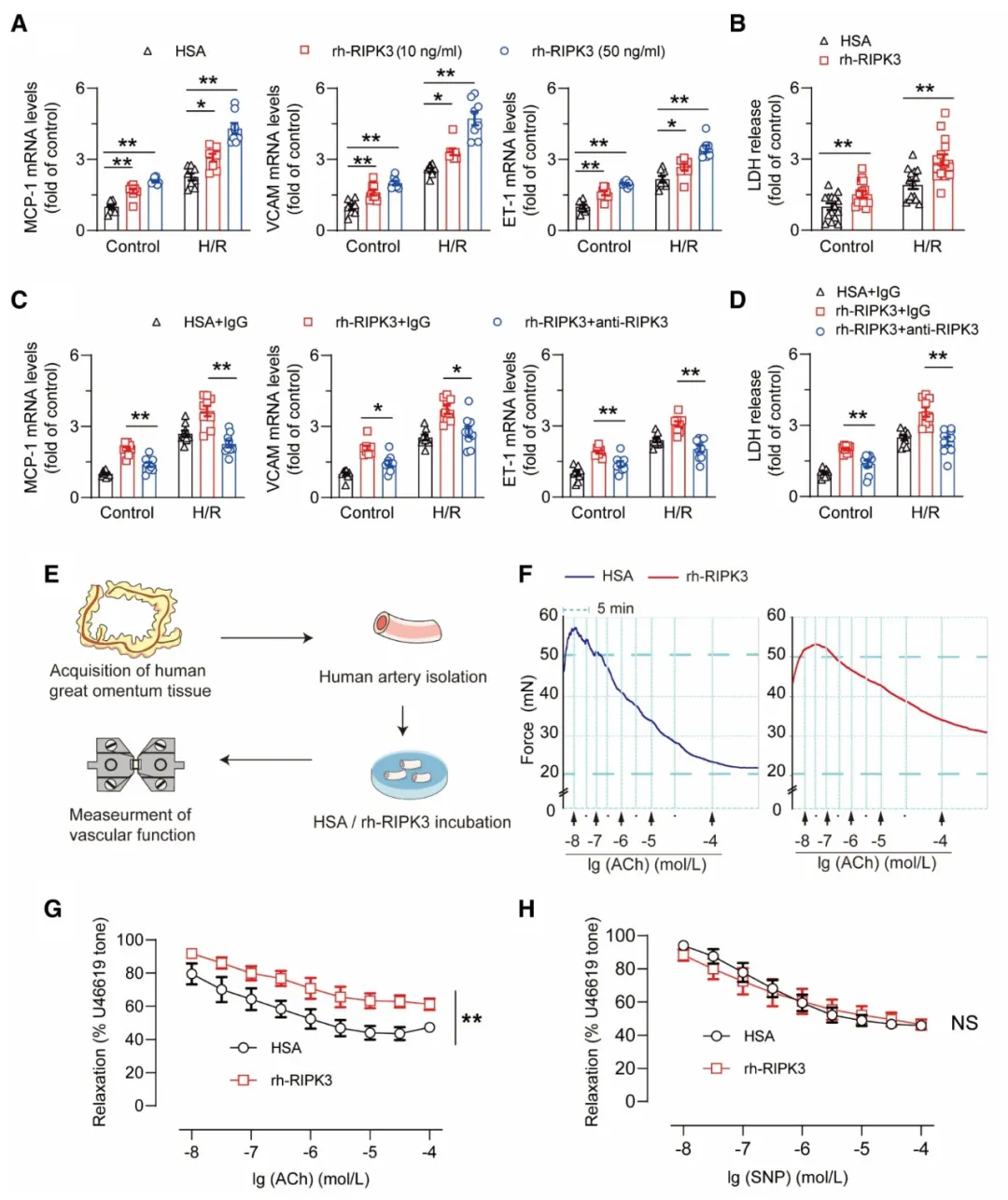
图4. 细胞外RIPK3增加了I/R诱导的内皮损伤。
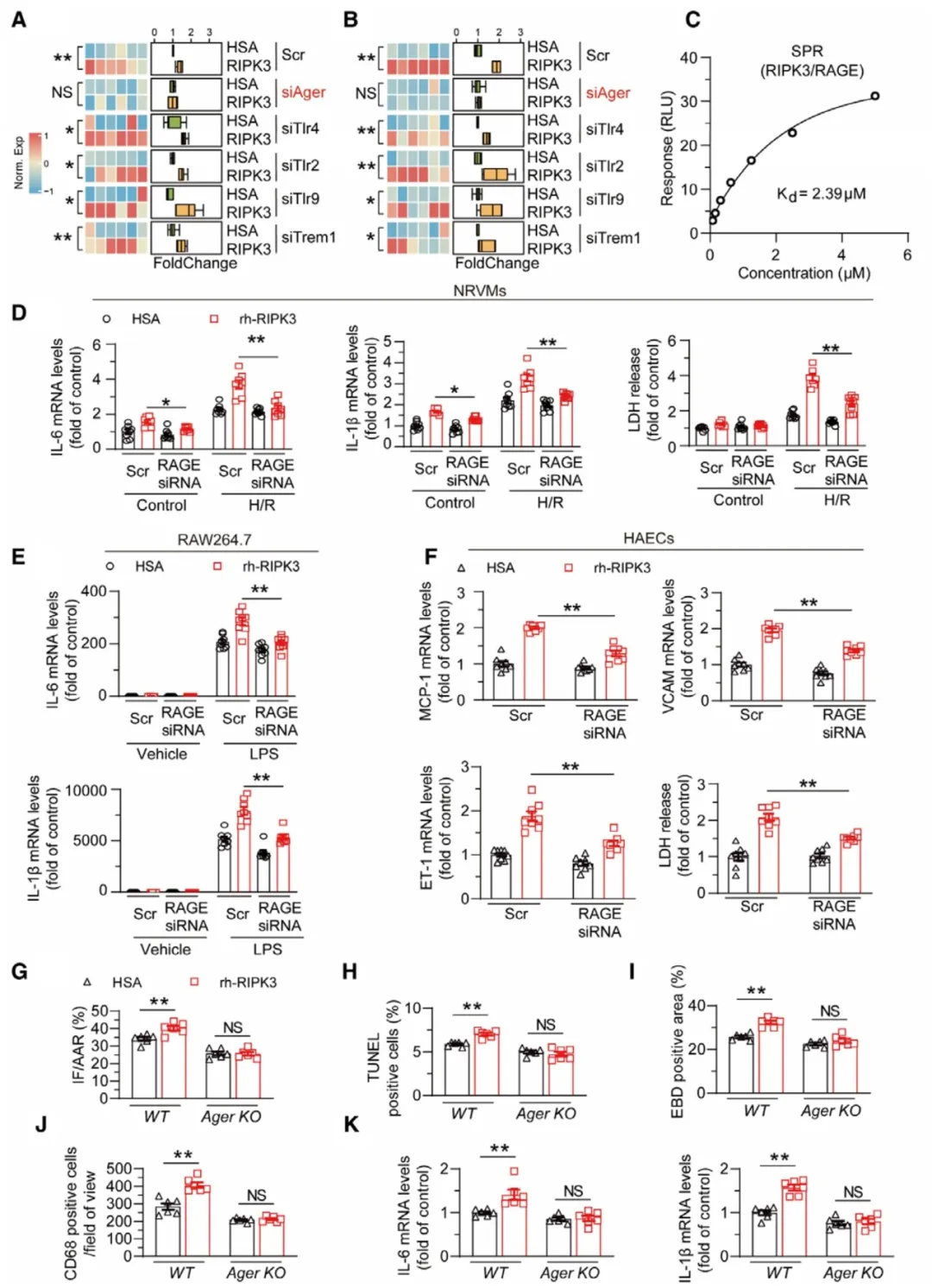
图5. 抑制RAGE减轻RIPK3诱导的炎症反应和心肌损伤。
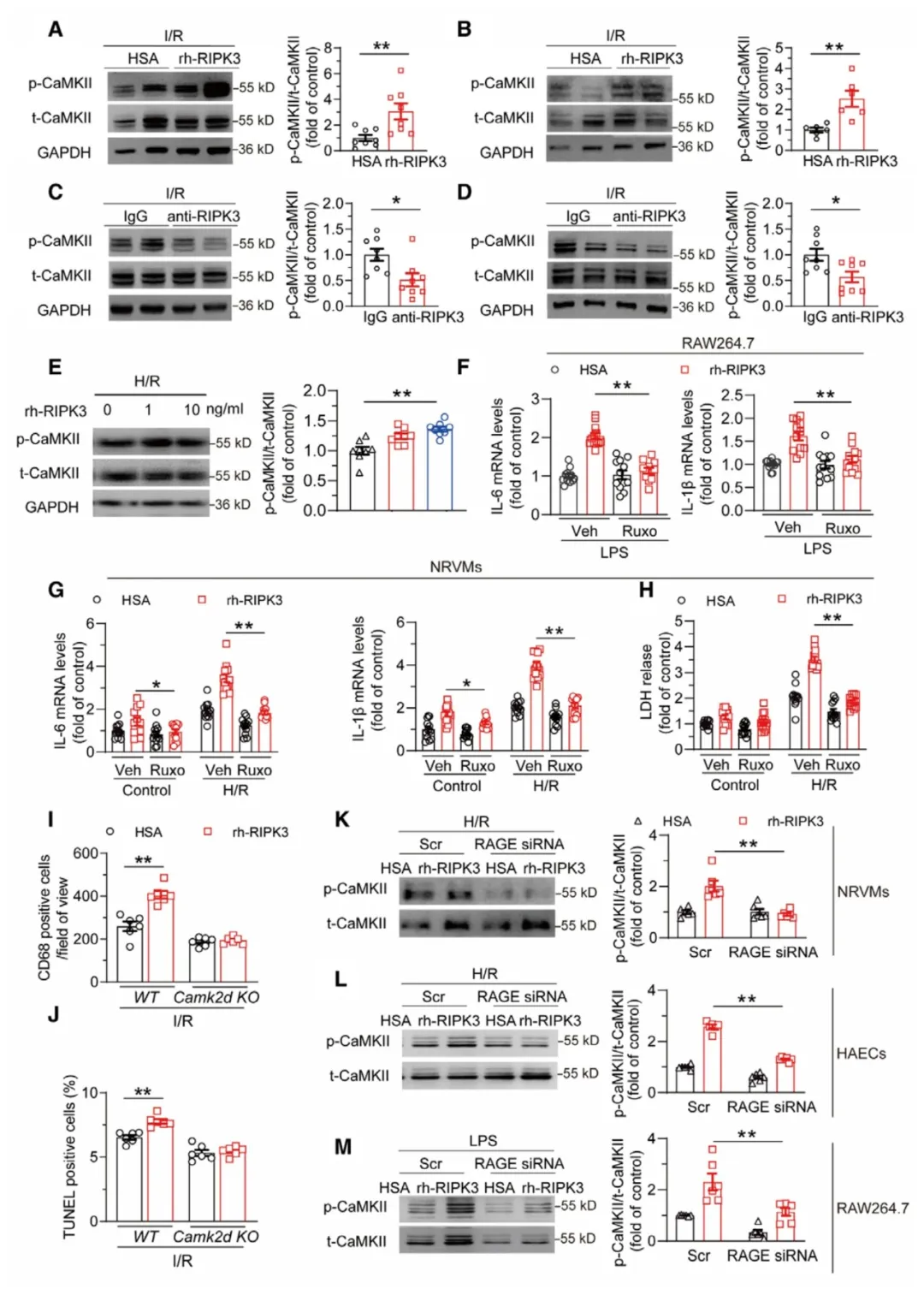
图6. CaMKII介导RIPK3诱导的细胞病理效应,位于RAGE下游。
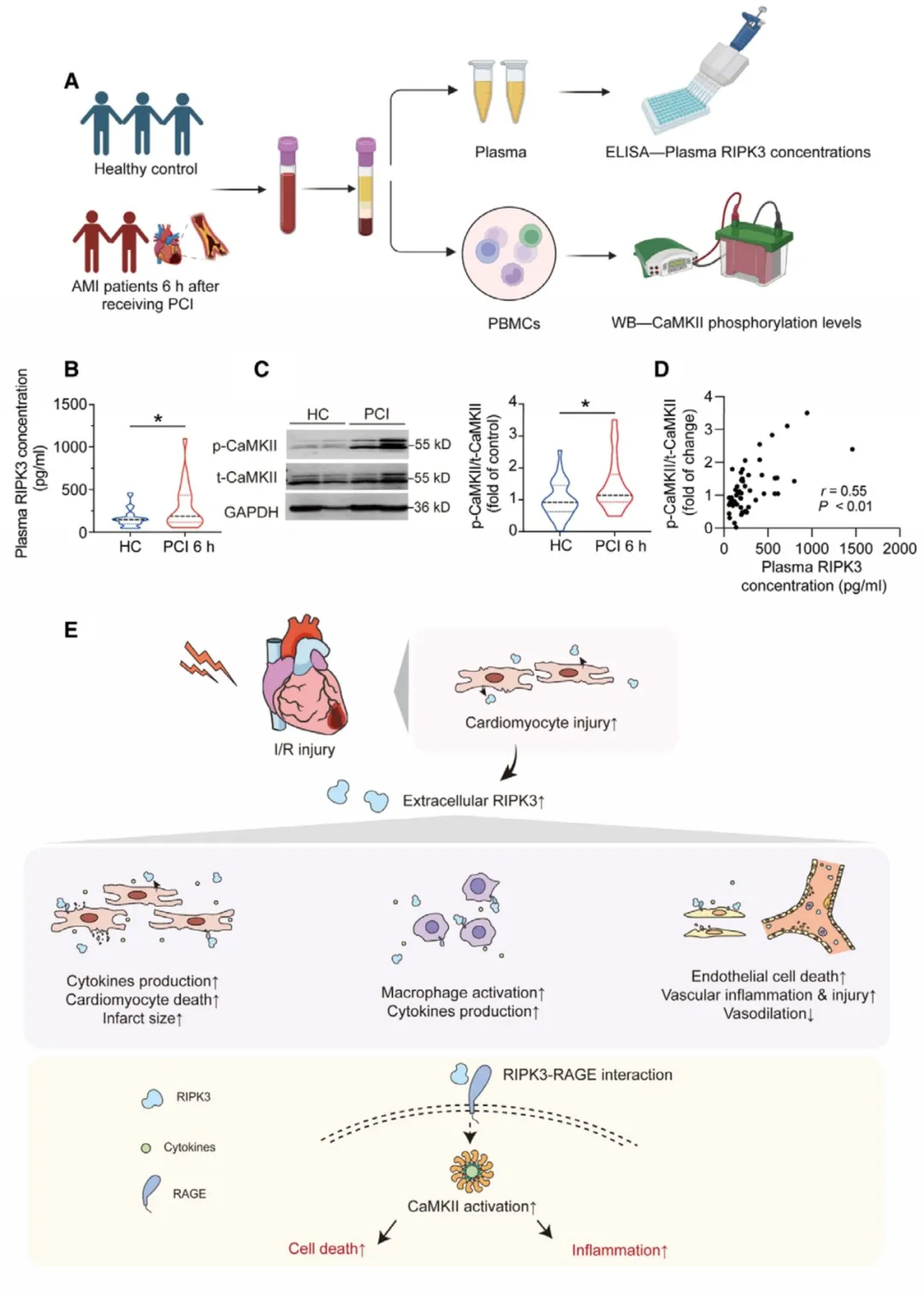
图7. 血浆RIPK3浓度与人类单核细胞中CaMKII磷酸化水平正相关。
结论与讨论
在接受PCI治疗的AMI患者中,血浆RIPK3水平的升高与不良心血管事件的风险增加有关。实验表明,细胞外RIPK3通过与RAGE结合激活CaMKII,进而促进心肌细胞死亡和炎症反应。这些结果揭示了RIPK3作为一个潜在的生物标志物和治疗靶点,用于改善心脏I/R损伤的临床结果。
尽管研究取得了初步成果,但血浆RIPK3作为生物标志物的诊断价值和阈值需要在更大规模的多中心随机临床试验中进一步验证。未来的研究还应包括对不同性别和种族的患者进行研究,以确保结果的普适性。此外,还需要进一步探索RIPK3/RAGE/CaMKII信号通路在其他心脏疾病中的作用,以及开发针对该通路的新型治疗策略。