















 {{item.name}}会员
{{item.name}}会员

Nature子刊 | 北京大学深圳医院发文揭示mTORC2介导的cGAS磷酸化在结直肠癌化疗敏感性调控中的关键作用
Highlights
1. mTORC2复合体通过在cGAS蛋白的丝氨酸37位点上直接磷酸化,促进cGAS与染色质的结合
2. ccGAS的缺失抑制了癌细胞的增殖,但也导致了对5-FU化疗药物的抗性
3. 提出通过靶向mTORC2-ccGAS轴和谷氨酰胺酶KGA来克服化疗抗性的可能性
近日,“Nature Cell Biology”(IF=17.3)发表了一篇题为“mTORC2-driven chromatin cGAS mediates chemoresistance through epigenetic reprogramming in colorectal cancer”的文章。这篇文章研究了在结直肠癌中,mTORC2复合体通过磷酸化cGAS蛋白的丝氨酸37位点,促进cGAS与染色质的结合,进而影响细胞生长和药物敏感性。

研究背景介绍
mTORC2是哺乳动物雷帕霉素靶蛋白(mTOR)的复合体之一,参与调节细胞生长、代谢和存活。
cGAS是一种胞质DNA感受器,能够识别细胞质中的双链DNA,并激活STING依赖的天然免疫反应。
SWI/SNF染色质重塑复合体是一种大型蛋白质复合体,通过改变染色质的结构来调节基因表达。它在多种生物学过程中发挥作用,包括细胞周期控制和癌症发展。
肾脏型谷氨酰胺酶(KGA)是谷氨酰胺代谢途径中的一种酶,它催化谷氨酰胺转化为α-酮戊二酸和氨,参与调节肿瘤细胞的代谢和生存。
研究思路分析
研究技术路线图01cGAS的染色质定位与mTORC2的调控
①高通量化合物筛选发现,PI3K–mTOR信号通路中的BEZ-235、GDC-0941和KU-0063794三种化合物能显著抑制cGAS与染色质的相互作用。且GDC-0941处理后的细胞中,cGAS和HELLS在染色质上的含量减少,且cGAS在染色质上的水平随处理时间增加而持续下降。shRNA敲低实验显示,敲减RICTOR抑制了cGAS与染色质的定位。BRET实验和染色质分馏实验进一步证实,敲减mTOR和RICTOR破坏了cGAS与染色质的相互作用。此外,酶活性失活的S213D cGAS突变体与染色质的相互作用也受mTORC2调控。表明cGAS与染色质结合依赖于mTORC2,而与其核苷酸转移酶活性无关。
②通过免疫共沉淀和BREY实验,发现cGAS与mTORC2的一个关键组分SIN1有相互作用。体外实验发现激活的mTORC2能够在cGAS的丝氨酸37位点进行磷酸化。在RICTOR缺失的细胞中,cGAS的丝氨酸37磷酸化水平降低,但可以通过RICTOR的重新引入得到恢复。GDC-0941处理会降低p-Ser37-cGAS的水平。免疫荧光分析显示p-Ser37-cGAS与组蛋白H2A在核内共定位,且仅染色质结合的cGAS发生Ser37磷酸化。S37D突变促进染色质结合,而S37A突变则破坏结合。mTORC2的抑制减少了Ser37磷酸化和cGAS的染色质定位,表明mTORC2介导的Ser37磷酸化对cGAS的染色质定位至关重要。
02ccGAS的功能与基因表达调控
①对ccGAS相互作用蛋白进行表征,发现ccGAS与核糖体亚单位在细胞核外部分相互作用,并招募了SWI/SNF染色质重塑复合体的多个组分,如SMARCC2、SMARCA4等,以调控基因表达。CUT&Tag实验表明ccGAS偏好结合富含G/A的基因转录起始位点附近区域,且与PI3K–AKT途径相关基因富集。通过TMT标记和定量蛋白质组学分析,发现ccGAS敲减后,与DNA复制相关的蛋白表达显著降低,如CDC45–MCM–GINS(CMG)解旋酶复合体的组分。表明ccGAS通过招募SWI/SNF到特定染色质区域,正向调控DNA复制蛋白的表达。
②通过蛋白质组学分析发现,ccGAS敲减的细胞中,有28种蛋白质显著上调,包括KGA。KGA在ccGAS敲减的结直肠癌细胞中的mRNA水平和谷氨酰胺酶活性显著增加,且这一现象可通过重新表达cGAS得到逆转,表明ccGAS负向调控KGA表达。CUT&Tag和ChIP实验证实ccGAS定位于KGA启动子区域,且这一定位受mTOR或RICTOR敲减的影响。GDC-0941处理增加了KGA的表达,与ccGAS蛋白水平降低相关。cGAS敲除细胞中KGA表达升高,但可通过S37D突变体重表达得到挽救。此外,cGAS敲除增加了谷氨酰胺依赖的氧气消耗,这一效应也可通过S37D突变体挽救,进一步证实ccGAS在调控KGA表达和谷氨酰胺分解中的作用。
03ccGAS与肿瘤生长、化疗抗性的关系
①ccGAS敲减导致HCT116细胞在nocodazole同步化后发生G1/S期阻滞,这一阻滞可通过过表达S37D或S213D cGAS突变体得到挽救,而S37A突变体或STING激活剂ADU-S100的补充则无效。平板克隆和细胞计数实验证实,ccGAS敲减抑制了HCT116细胞生长,且S37D过表达可挽救这一抑制。ccGAS定位于染色质的细胞表现出小的梭形“上皮样”形态。cGAS敲除细胞中S37D突变体表达呈现间充质特征,而S37A突变体则呈现滞育样形态。抑制PI3K–mTORC2或RICTOR敲减诱导了滞育样形态,而抑制AKT1–mTORC1–S6K轴或RAPTOR敲减则不影响细胞形态。表明,抑制mTORC2–ccGAS轴诱导了结直肠癌细胞滞育样可塑性。
②研究发现,ccGAS敲减抑制了HCT116细胞的增殖,但增加了对5-FU诱导的死亡抵抗。野生型ccGAS的重表达或S37D和S213D突变体的过表达可恢复对5-FU的敏感性,而S37A突变体无效。此外,STING激活或敲减不影响结直肠癌细胞对5-FU的抵抗。ccGAS调控的化学抗性独立于MLH1状态,并广泛适用于具有不同微卫星不稳定性的结直肠癌细胞系。在体内小鼠异种移植模型中,ccGAS敲减显著抑制了肿瘤的形成和生长,而S37D突变体的过表达可恢复这些效果。在5-FU治疗下,ccGAS敲减的肿瘤生长速度较慢,对5-FU的响应性降低,而S37D突变体的过表达恢复了5-FU的响应性。表明,即使ccGAS的缺失降低了增殖能力,它也促进了化学抗性,恢复ccGAS功能可能克服化学抗性。
③在患者衍生异种移植模型中,RICTOR缺失肿瘤的生长速度显著减慢。5-FU治疗显著减少了野生型对照小鼠的肿瘤数量和大小,但对RICTOR缺失小鼠没有效果。然而,联合5-FU和BPTES显著减轻了RICTOR缺失小鼠的肿瘤负担。表明,抑制KGA可以克服cGAS缺失诱导的化学抗性,并且针对ccGAS和KGA的靶向可能代表了结直肠癌治疗的一个有前景的策略。
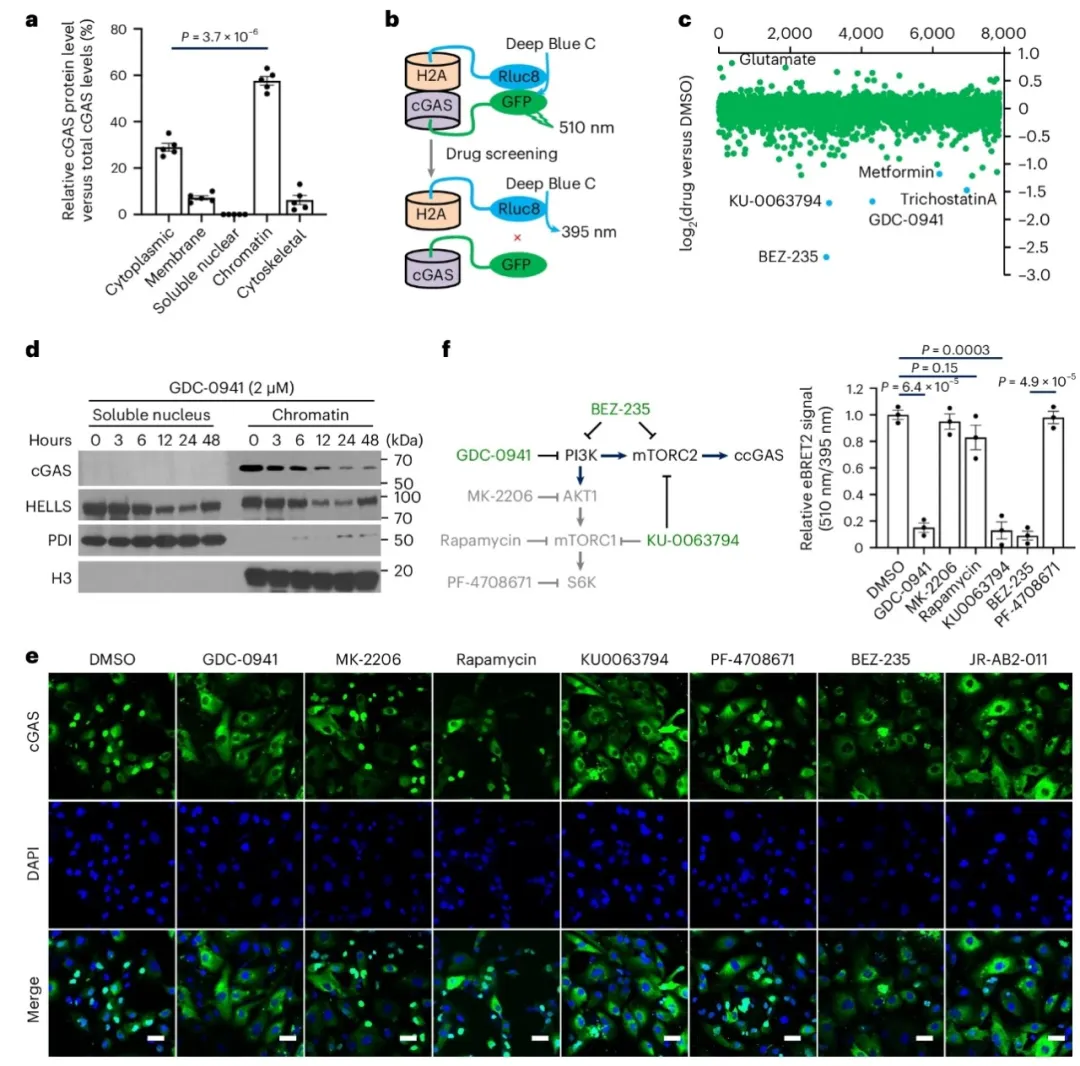
图1. 高通量筛选可识别cGAS-染色质定位的PI3K-mTOR通路调控
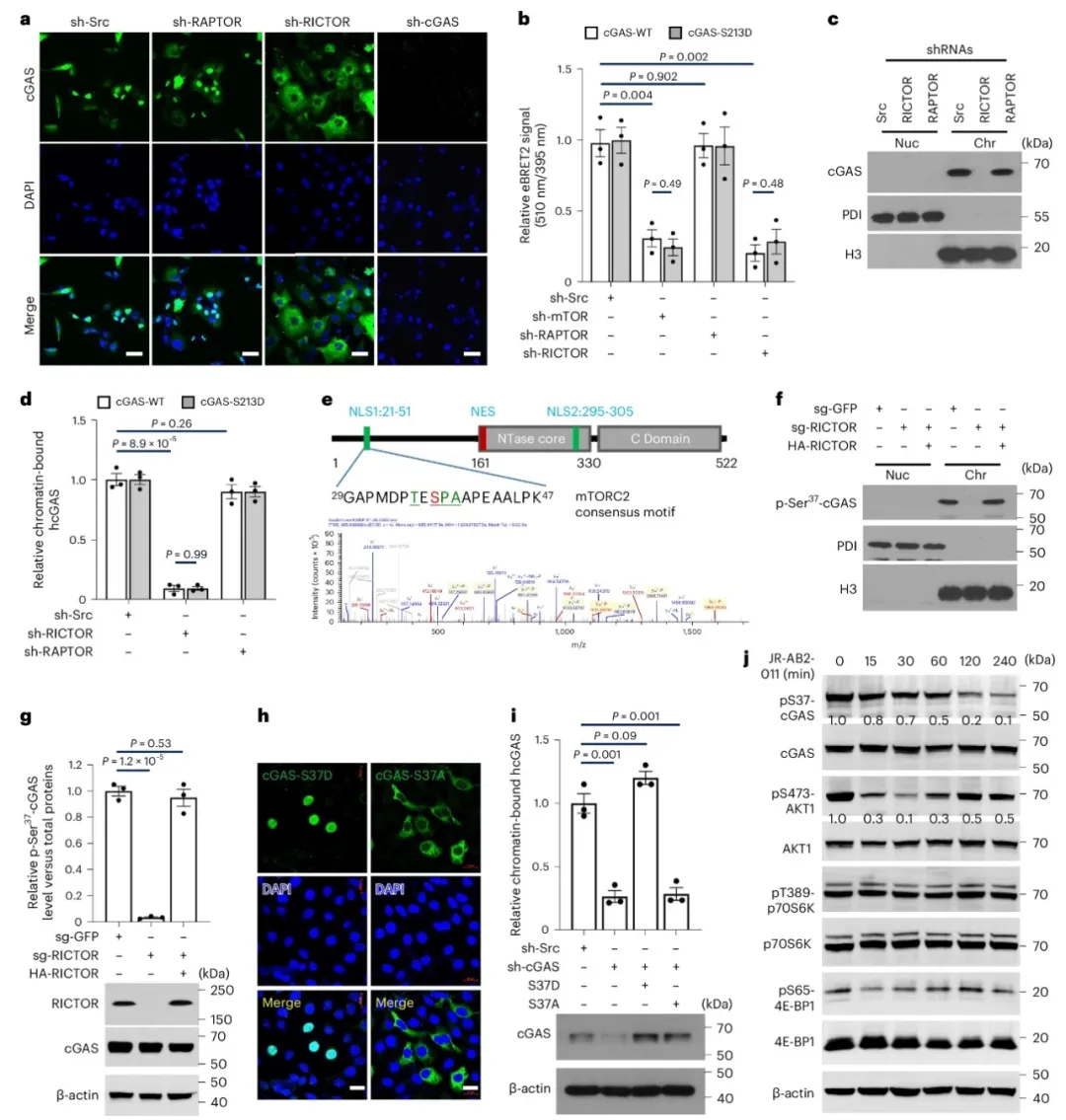
图2. mTORC2 介导的 cGAS 丝氨酸 37 磷酸化促进其染色质定位
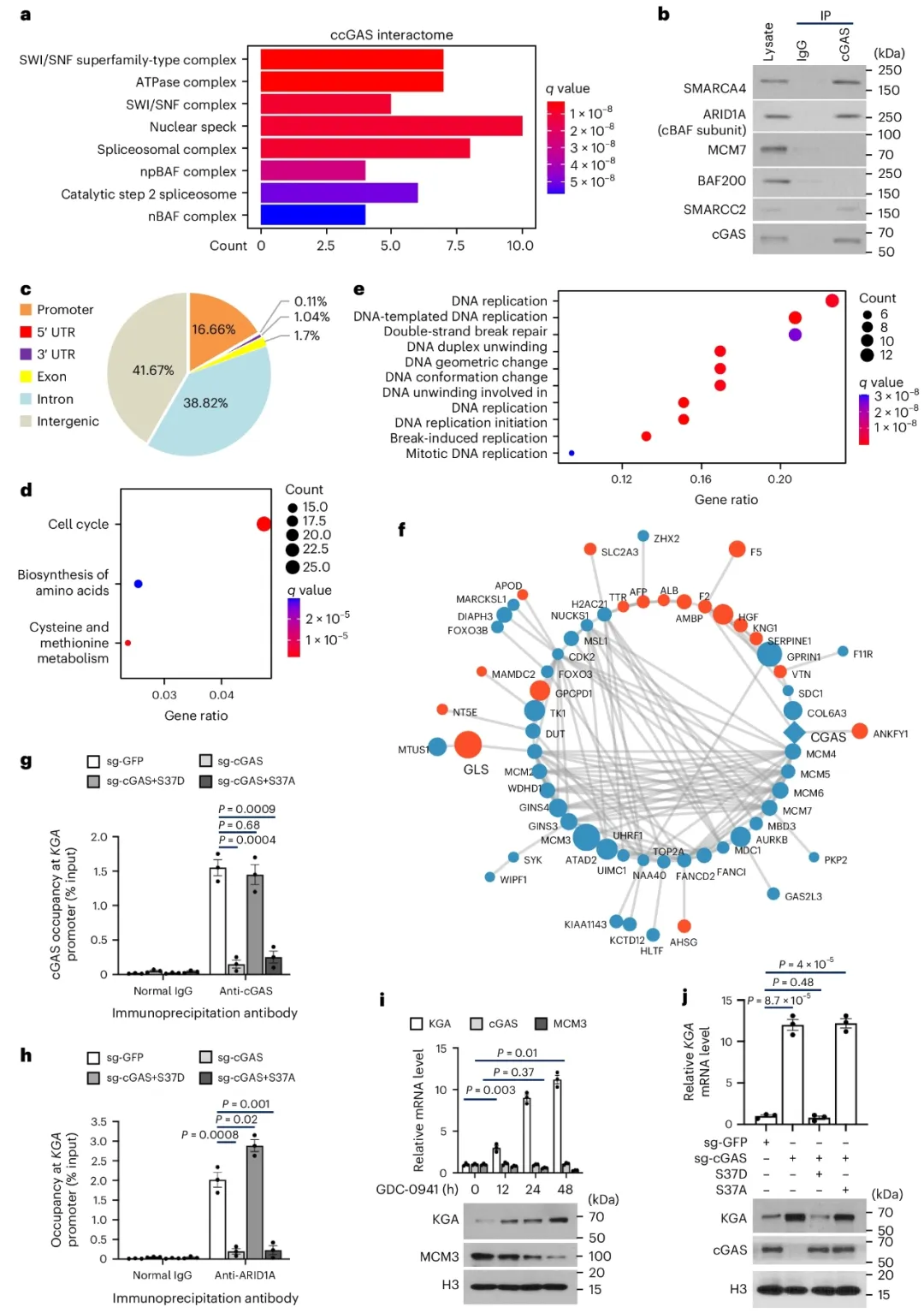
图3. ccGAS募集SWI/SNF来调节参与DNA复制和谷氨酰胺分解的基因
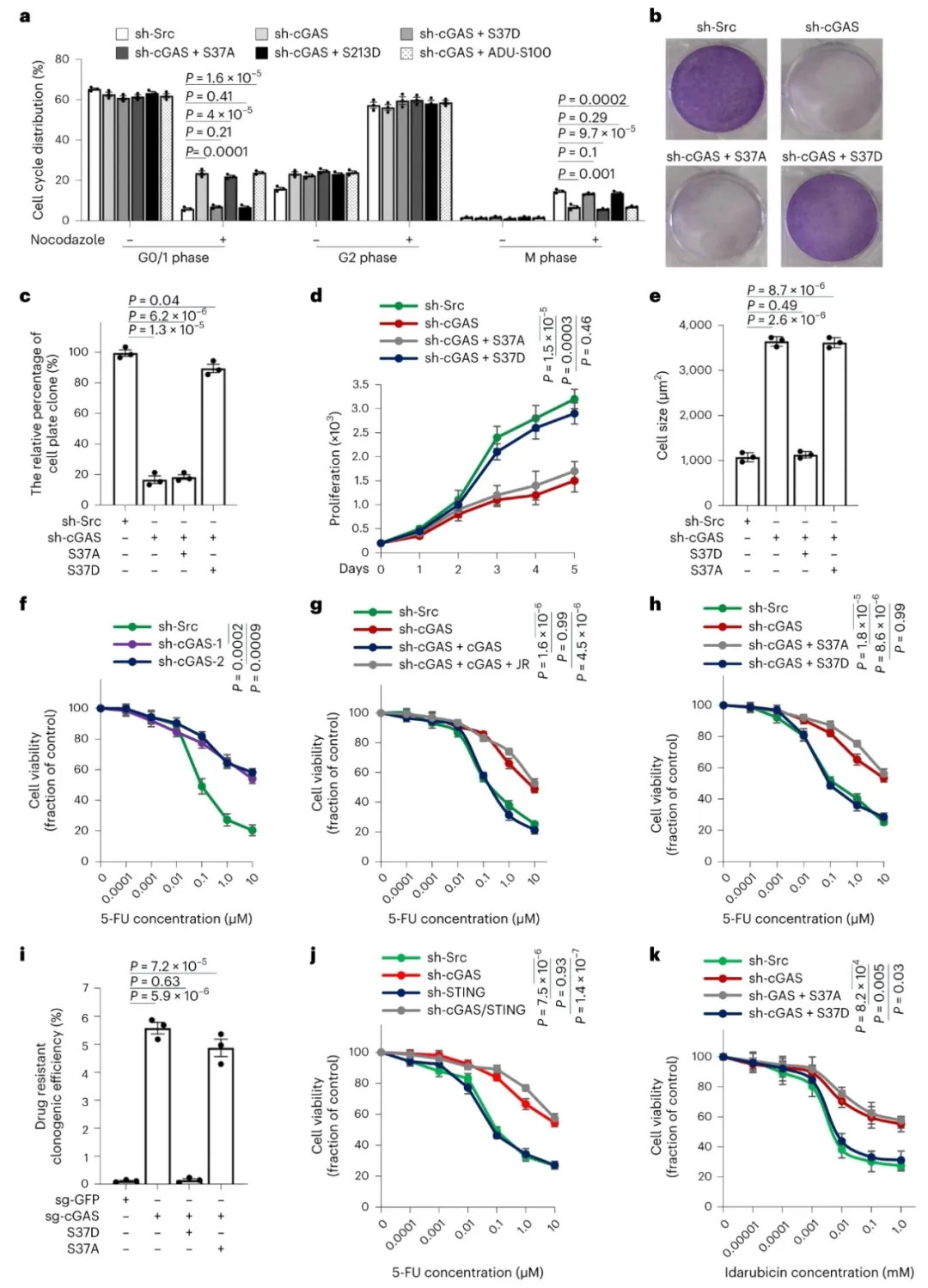
图4. ccGAS耗竭诱导结直肠癌细胞出现滞育样状态和化疗耐药性
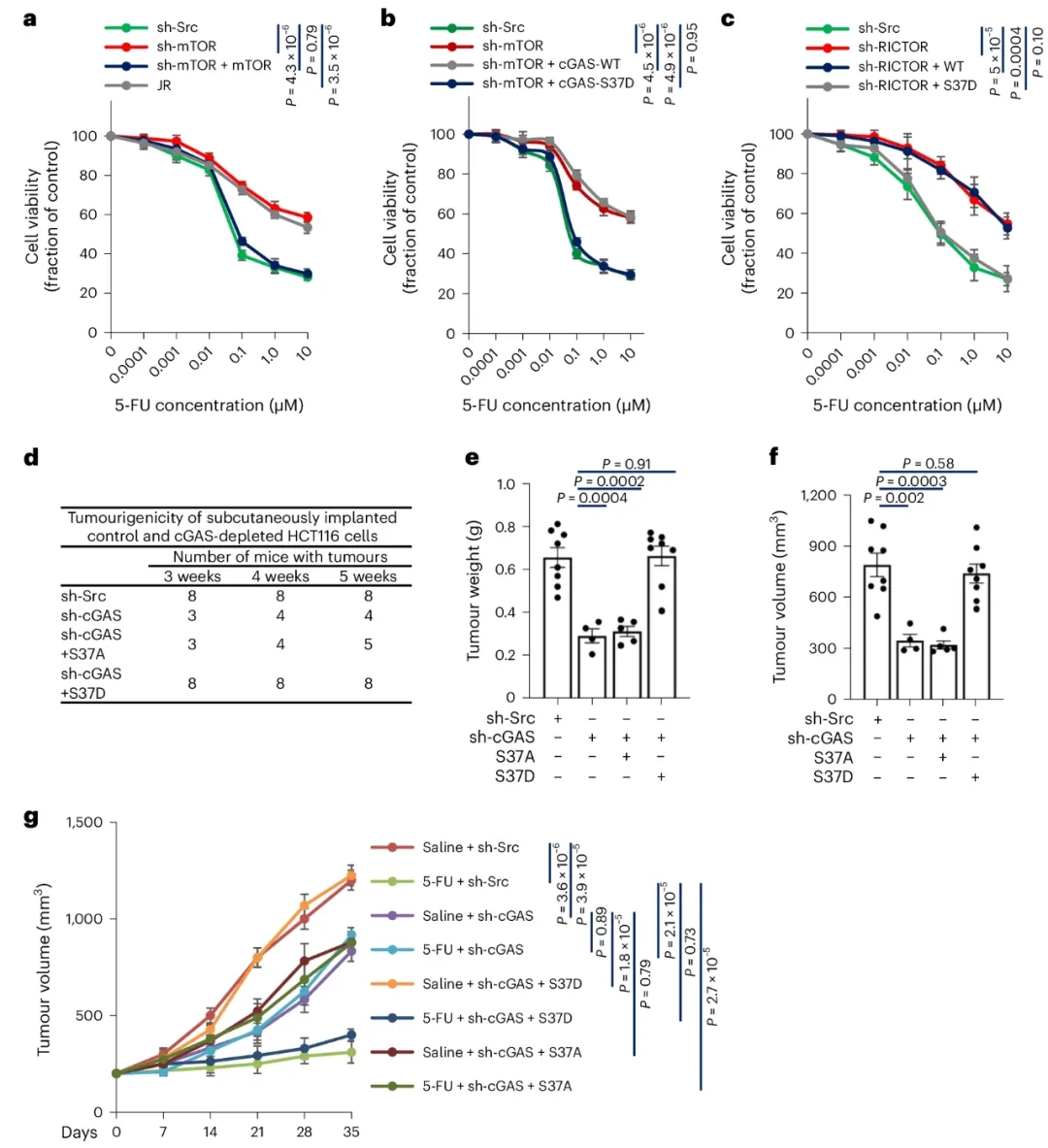
图5. mTORC2 驱动的 ccGAS 在体内指导结直肠癌可塑性和获得性化疗耐药性
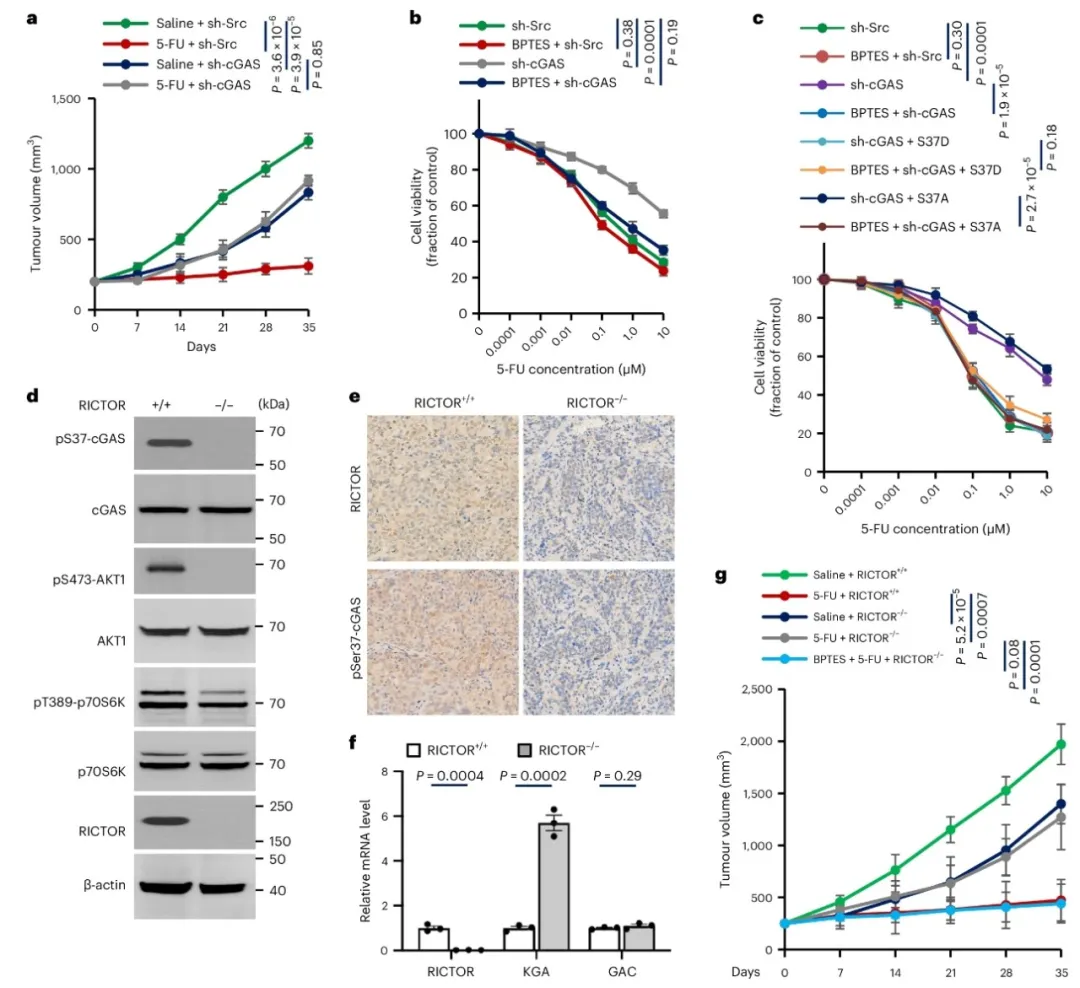
图6. KGA抑制克服了由mTORC2-ccGAS轴破坏诱导的化学耐药性
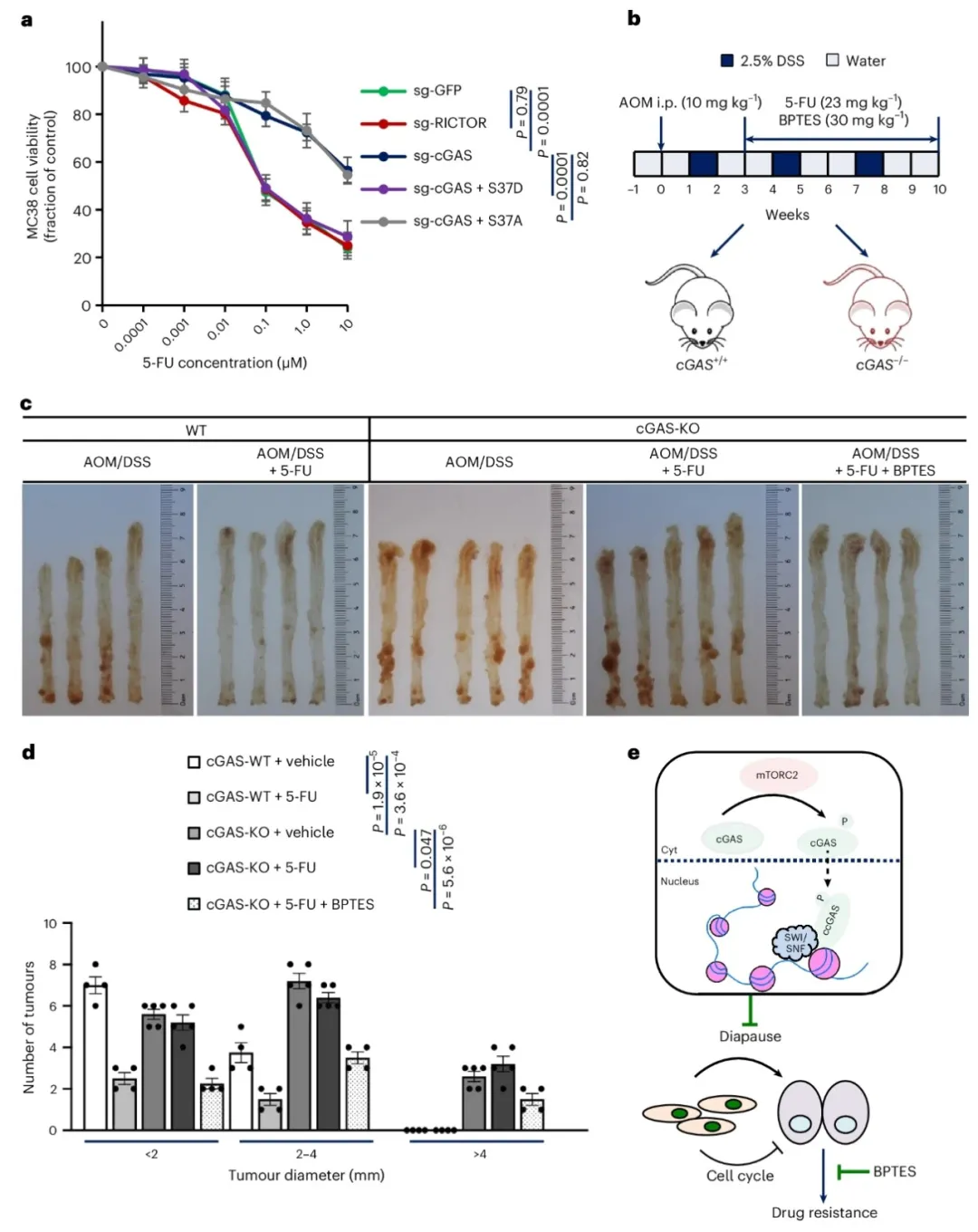
图7. 靶向KGA在cGAS缺陷小鼠的肿瘤中重新建立化疗敏感性
结论与讨论
研究发现mTORC2通过在丝氨酸37位点磷酸化cGAS,促进其在结直肠癌细胞中的染色质定位,独立于STING调控细胞生长和药物抗性。ccGAS的缺失虽然抑制了细胞增殖,却诱导了对5-FU的化疗抗性。研究还表明,通过靶向KGA,可以克服由ccGAS缺失引起的化疗抗性,为结直肠癌治疗提供了新的策略。
然而,尽管已揭示mTORC2-ccGAS-KGA轴在细胞可塑性和获得性化疗抗性中的作用,但仍需在更多模型和临床环境中验证这些发现。未来研究应探索mTORC2介导的S37磷酸化之外的其他调控因素,并利用多组学数据和纵向分析来加强这些发现的转化相关性。